Handle gem files for downstream analysis
GEM is a tabular structure format specified for saving stereo-seq data, which generally contains columns of x, y, geneID, MIDCount and decodes the feature information captured by each DNB on the stereo-seq chip. Additional columns counld be included for DNB/feature annotation. A demo description can be found at stereopy site.
[1]:
import sys
sys.path.append('../../')
[2]:
from spacipy import Gem
Read in example gem data, which holds mouse brain stereo-seq data from here.
[3]:
gem = Gem.readin('../_static/data/example.gem.gz')
[4]:
gem
[4]:
| geneID | x | y | MIDCounts | |
|---|---|---|---|---|
| 0 | Cr2 | 10059 | 14612 | 1 |
| 1 | Cr2 | 6066 | 11227 | 3 |
| 2 | Cr2 | 10521 | 11108 | 1 |
| 3 | Cr2 | 8072 | 11782 | 3 |
| 4 | Cr2 | 7891 | 16372 | 1 |
| ... | ... | ... | ... | ... |
| 106663420 | CAAA01147332.1 | 12756 | 17013 | 1 |
| 106663421 | CAAA01147332.1 | 11907 | 12871 | 1 |
| 106663422 | CAAA01147332.1 | 14707 | 4862 | 1 |
| 106663423 | CAAA01147332.1 | 9787 | 5437 | 1 |
| 106663424 | CAAA01147332.1 | 10471 | 15907 | 1 |
106663425 rows × 4 columns
preliminary understanding of your data
GEM saves spatial transcriptomics data, which can be easily understanding by seeing what it looks like.
[5]:
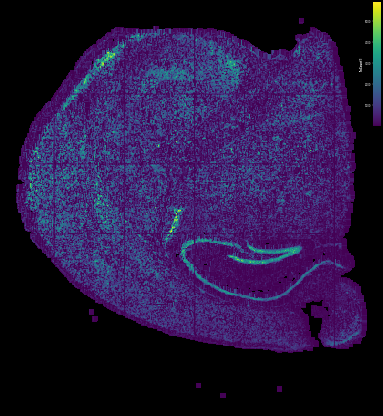
gem.plot(color='nCount', bin_size=20)
None
[5]:
[<Axes: >]

here you may focus on two points: 1. whether the spatial distributuion of the data is well decoded 2. how many features were captured in each square bin or DNB
extract data where tissue covered
Not all DNBs on the stereo-seq chip were covered by the tissue. An effective way for deleting non-related data is to use a mask, which is often a two dimensional numpy ndarray with tissue regions labeled as non-zero integer (see here). Once prepared a ready-to-use mask matrix, you can process as following:
[6]:
import numpy as np
mask = np.loadtxt('../_static/data/example.transformed_mask.txt.gz')
mask.shape
[6]:
(14451, 14248)
[7]:
gem.img_shape
[7]:
(14451, 14248)
[8]:
gem = gem.mask(matrix=mask, return_offset=False, label_object=True)
now the gem data will be
[9]:
gem.plot(color='nCount', bin_size=20)
None
[9]:
[<Axes: >]
aggregate the data into cell/bin unit
After getting the effective data set, the next step is to determine how to group the nanometer-resolution DNB array into a analysis-ready unit, here always in two way: 1. group into square bin with equal width and height 2. group into putative cells
1. group into square bin
Square binning strategy do NOT consider any other informations like nucleus location and molecular homogeny. The only parameter in this step is bin_size, which specify how many DNBs in width and height should be grouped as a single bin unit. People can change this value based on the physical area of each bin and the captured features in each bin unit.
[10]:
binning_gem = gem.binning(bin_size=20)
binning_gem
[10]:
| geneID | x | y | label | MIDCounts | |
|---|---|---|---|---|---|
| 0 | 0610005C13Rik | 5780 | 12540 | 16200 | 3 |
| 1 | 0610005C13Rik | 5960 | 12760 | 14612 | 1 |
| 2 | 0610005C13Rik | 5980 | 10700 | 28985 | 3 |
| 3 | 0610005C13Rik | 6100 | 10240 | 32093 | 1 |
| 4 | 0610005C13Rik | 6140 | 11420 | 23757 | 2 |
| ... | ... | ... | ... | ... | ... |
| 31798037 | mt-Nd6 | 12880 | 8940 | 37309 | 2 |
| 31798038 | mt-Nd6 | 12880 | 8960 | 37122 | 2 |
| 31798039 | mt-Nd6 | 12880 | 9140 | 36185 | 1 |
| 31798040 | mt-Nd6 | 12900 | 8640 | 39087 | 2 |
| 31798041 | mt-Nd6 | 12900 | 8840 | 37995 | 3 |
31798042 rows × 5 columns
2. group into putative cells
Currently stereo-seq comprehend ssDNA stainning protocal, which stains the nucleus of cells on the same section. With the location of cell nucleus decoded, labeled with numpy mask file. And in the previous gem.mask process, we have labeled each DNB with their cell IDs by setting label_object=True.
[11]:
gem
[11]:
| geneID | x | y | MIDCounts | label | |
|---|---|---|---|---|---|
| 0 | Cr2 | 8072 | 11782 | 3 | 20638 |
| 1 | Cr2 | 7832 | 10872 | 1 | 27089 |
| 2 | Cr2 | 6608 | 10674 | 1 | 28950 |
| 3 | Cr2 | 9790 | 14297 | 2 | 3574 |
| 4 | Cr2 | 6594 | 12299 | 2 | 17562 |
| ... | ... | ... | ... | ... | ... |
| 37759281 | CAAA01147332.1 | 12231 | 11534 | 1 | 20817 |
| 37759282 | CAAA01147332.1 | 10776 | 12505 | 2 | 14553 |
| 37759283 | CAAA01147332.1 | 9810 | 10370 | 1 | 29920 |
| 37759284 | CAAA01147332.1 | 10653 | 12974 | 1 | 11713 |
| 37759285 | CAAA01147332.1 | 8062 | 11252 | 2 | 24214 |
37759286 rows × 5 columns
with cell ID labels, the gem file could be aggregated into cell-gene matrix as AnnData object
[13]:
adata = gem.to_anndata()
adata
[13]:
AnnData object with n_obs × n_vars = 41502 × 25288
the anndata object can be write into h5ad file and further used in clustering analysis
[15]:
adata.write('../_static/data/example.h5ad')